Computational modelling in chemistry and materials science
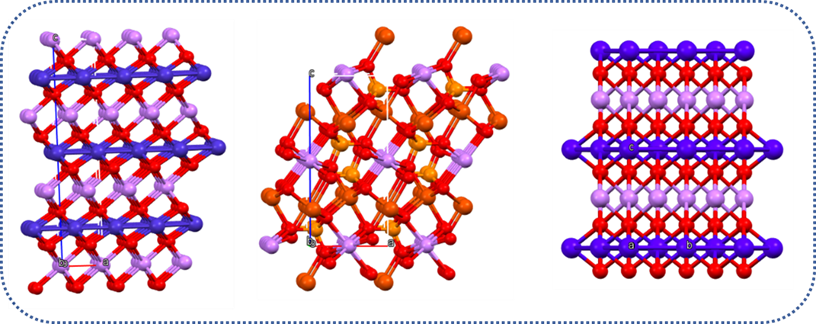
We use density functional theory (DFT) based computer simulations to study systems of varying size (molecules, clusters, nano-structures, surfaces and bulk materials) and probe their structure and stability, electronic properties and chemical reactions. We work in close collaborations with experimental groups to understand the underlying chemical phenomena responsible for the experimental observations.
Focus of our present computational research
- Computational studies of important battery properties
- Investigations for next-generation efficient battery materials through design and modelling
- Designing catalyst materials for clean energy